遗传性儿童肾脏疾病背后的一个新基因已被一个全球性研究团队确定。 昆士兰大学的研究人员是这个研究团队的一部分,他们的发现将改进基因检测的方式,并为未来常染色体隐性多囊肾病(ARPKD)的治疗提供线索。
UQ研究所分子生物科学研究所研究员副研究员Carol Wicking是该研究的主要作者之一,他说以前很难确定所有ARPKD病例的根本原因。
Wicking副教授说:“先前的研究表明,这种被称为PKHD1的基因如果发生错误,就会导致一种罕见的肾脏疾病。但是,似乎总是存在一部分患有这种疾病的患者,即使他们拥有正常的基因,但仍会患病。”
这项研究的目的是找到对这种破坏性状况负责的其他遗传罪犯。ARPKD疾病会导致肾脏扩大、肝脏问题和高血压,并且70%的患者在生命的头几周经常会肾衰竭而致命。
使用称为整体外显子测序技术同时分析患者的所有基因,德国和美国的研究人员发现在具有ARPKD的四个家族中被称为DZIP1L的基因中存在错误。
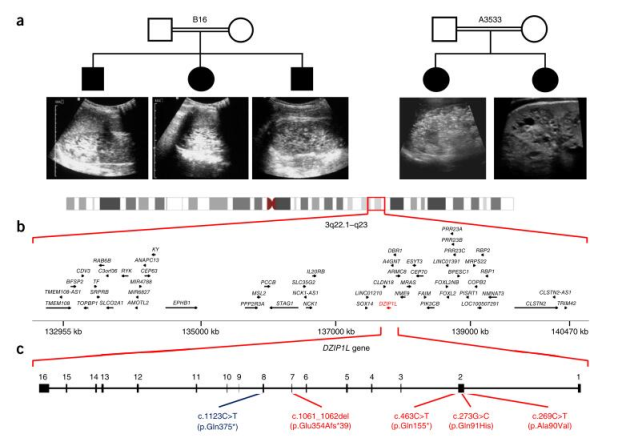
通过在小鼠和斑马鱼模型上的进一步验证,澳大利亚,新加坡和德国的Wicking副教授及其同事使用基于实验室的模型来确认该基因的错误确实会导致肾脏缺陷,并开始探索并试图理解其中的原因。
研究发现DZIP1L位于中心粒和基底体的远端,并与septin2(SEPT2)相互作用,septin2(SEPT2)是参与过渡区扩散屏障维持的蛋白质。与这些数据一致的是,研究发现在DZIP1L缺陷型原发性纤毛中,PC1和PC2显示沿睫状膜分布改变。
“DZIP1L基因似乎与纤毛的功能有关,它们几乎在所有细胞(包括肾脏中的细胞)中以一种小型天线般的延伸方式存在着,并且在控制重要的细胞功能中发挥重要作用。” Wicking教授说。
“这种基因使蛋白质在纤毛的基部发挥作用,当有缺陷时,会引起多米诺骨牌效应,从而导致纤毛问题,反过来又是一种功能障碍的肾脏。ARPKD的原因比原来想象的更复杂,我们了解这种罕见疾病的工作可能最终有助于我们更好地管理罕见和更常见的多囊肾病。”
许多患有罕见疾病的患者等待几年进行遗传诊断,并经常忍受多种误诊。澳大利亚罕见声音执行总监尼科尔・米利斯(Nicole Millis)表示,这一令人兴奋的发现为患者提供了急需的答案。
“这些研究结果突显了新型基因组技术如何有助于找到有罕见疾病患者的答案,使他们更加确定他们的病情。Millis女士说:“进行遗传诊断也使患者及其家属有机会与其他生活在类似稀有疾病的人建立联系并建立重要的支持网络。
|